The approval of new drugs in India is regulated by the Central Drugs Standard Control Organization (CDSCO) under the leadership of the Central Drugs Standard Control Organization and the Drugs Controller General of India (DCGI). Any pharmaceutical company planning to manufacture, import, market, or conduct clinical trials for a new drug must obtain regulatory approval from CDSCO before commercialization. The approval process is primarily governed by the New Drugs and Clinical Trials (NDCT) Rules, 2019.
What is CDSCO?
The Central Drugs Standard Control Organization is India’s national regulatory authority responsible for:
- Approval of new drugs
- Clinical trial permissions
- Import and export licenses
- Medical device regulation
- Pharmacovigilance oversight
- Ensuring drug safety, efficacy, and quality
CDSCO functions under the Ministry of Health & Family Welfare, Government of India.
What is Considered a “New Drug”?
According to NDCT Rules, 2019, a drug may be classified as a “new drug” if it:
- Contains a new active pharmaceutical ingredient (API)
- Uses a new dosage form or route of administration
- Has a new indication
- Is a biological product, vaccine, monoclonal antibody, gene therapy, or cell-based therapy
- Has not previously been approved in India
Many approved drugs continue to be treated as “new drugs” for a specified period after approval.

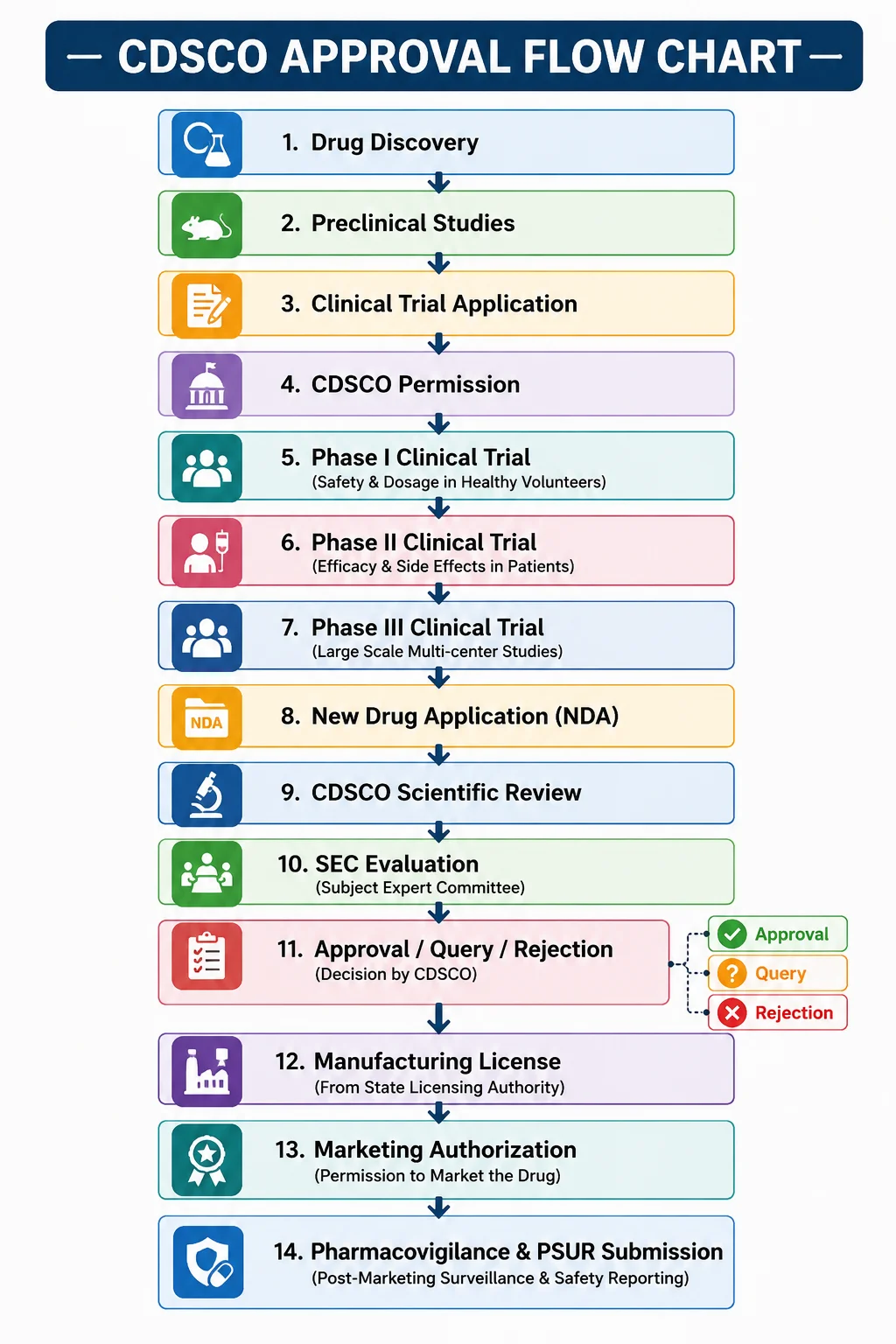
Step-by-Step CDSCO Approval Process
Step 1: Drug Development & Preclinical Studies
Before approaching CDSCO, the sponsor conducts:
Laboratory Studies
- Pharmacological studies
- Toxicological studies
- Stability studies
Animal Studies
- Acute toxicity
- Chronic toxicity
- Reproductive toxicity
- Carcinogenicity studies
The objective is to generate sufficient safety data before human testing begins.
Step 2: Clinical Trial Application
If the drug requires clinical evaluation in humans, the company submits an application to CDSCO through the SUGAM portal.
Documents generally include:
- Preclinical study reports
- Investigator’s Brochure
- Clinical Trial Protocol
- Ethics Committee approvals
- Informed Consent Documents
- Manufacturing information
CDSCO reviews the submitted data before granting permission to conduct clinical trials.
Step 3: Clinical Trial Phases
Phase I
- Conducted on a small group of healthy volunteers.
- Evaluates safety and dosage.
Phase II
- Conducted on patients.
- Assesses efficacy and side effects.
Phase III
- Large-scale studies involving multiple centers.
- Confirms safety and effectiveness.
Phase IV
- Post-marketing surveillance after approval.
Clinical trial requirements may vary depending on whether the drug is developed in India or already approved in other countries.
Step 4: Submission of New Drug Application (NDA)
After successful clinical trials, the sponsor submits a New Drug Application to CDSCO.
The application generally contains:
Quality (CMC) Data
- Manufacturing process
- Specifications
- Validation reports
- Stability data
Non-Clinical Data
- Toxicology reports
- Pharmacology studies
Clinical Data
- Clinical trial reports
- Statistical analyses
- Safety summaries
Additional Documents
- GMP certificates
- Product labeling
- Package insert
- Risk management plans

Step 5: Technical Review by CDSCO
CDSCO experts evaluate:
- Safety profile
- Efficacy data
- Quality standards
- Benefit-risk assessment
- Manufacturing compliance
During review, CDSCO may issue:
- Queries
- Deficiency letters
- Requests for additional data
The applicant must respond within the stipulated timeline.
Step 6: Subject Expert Committee (SEC) Review
For many applications, the dossier is referred to a Subject Expert Committee.
The SEC:
- Reviews scientific evidence
- Evaluates clinical trial outcomes
- Assesses risk-benefit ratio
- Recommends approval, rejection, or additional studies
Expert committee recommendations play a crucial role in the final decision.
Step 7: CDSCO Approval Decision
After reviewing all scientific and regulatory data, CDSCO may:
Approve the Drug
Permission is granted for manufacturing, import, sale, or distribution.
Request Additional Information
The applicant may need to submit more data.
Reject the Application
If safety, efficacy, or quality standards are not adequately demonstrated.
Step 8: Manufacturing License
Once CDSCO approval is obtained, manufacturers must secure the appropriate manufacturing license from the respective State Licensing Authority.
Requirements include:
- GMP-compliant facilities
- Qualified technical staff
- Quality control systems
- Regulatory documentation
Step 9: Post-Marketing Surveillance (Pharmacovigilance)
Approval does not end regulatory oversight.
Companies must:
- Monitor adverse drug reactions (ADRs)
- Submit Periodic Safety Update Reports (PSURs)
- Maintain pharmacovigilance systems
- Report serious adverse events promptly
CDSCO can inspect, suspend, or withdraw approvals if safety concerns arise
Common Reasons for Approval Delays
- Incomplete dossiers
- Insufficient clinical data
- Manufacturing deficiencies
- Poor quality documentation
- Delayed responses to CDSCO queries
- Inadequate pharmacovigilance plans
Proper regulatory planning can significantly reduce approval timelines.
Recent Regulatory Reforms
The Government of India has introduced amendments to the NDCT Rules to simplify procedures, reduce approval timelines, and promote innovation in drug development. CDSCO has also initiated measures to accelerate regulatory reviews and strengthen scientific evaluation capacity.
Conclusion
The CDSCO approval process is a comprehensive regulatory pathway designed to ensure that medicines entering the Indian market are safe, effective, and of high quality. From preclinical research and clinical trials to marketing authorization and pharmacovigilance, every stage requires strict compliance with regulatory requirements. Understanding the CDSCO approval framework helps pharmaceutical companies, regulatory affairs professionals, and aspiring Drug Inspectors navigate India’s evolving drug regulatory landscape efficiently.
Recommended Products